"SYMPTOMS"
Tay–Sachs disease is typically first noticed in infants around 6 months old displaying an abnormally strong response
to sudden noises or other stimuli, known as the "startle response". There may also be listlessness or muscle stiffness (hypertonia). The disease is classified into several forms, which are differentiated based on the onset age of neurological symptoms.
"PREVENTION"
Three main approaches have been used to prevent or reduce the incidence of Tay–Sachs:
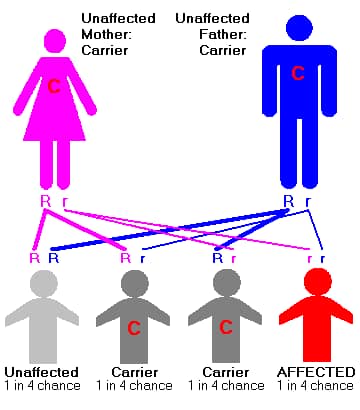
Prenatal diagnosis. If both parents are identified as carriers, prenatal genetic testing can determine whether the
fetus has inherited a defective gene copy from both parents. Chorionic villus sampling (CVS), the most common
form of prenatal diagnosis, can be performed between 10 and 14 weeks of gestation. Amniocentesis is usually performed at 15–18 weeks.
These procedures have risks of miscarriage of 1% or less.
Preimplantation genetic diagnosis. By retrieving the mother's eggs for in vitro fertilization, it is possible to test the
embryo for the disorder prior to implantation. Healthy embryos are then selected and transferred into the mother's womb, while unhealthy
embryos are discarded. In addition to Tay–Sachs disease, preimplantation genetic diagnosis has been used to prevent cystic fibrosis and
sickle cell anemia among other genetic disorders.
Mate selection. In Orthodox Jewish circles, the organization Dor Yeshorim carries out an anonymous screening program so that
carriers for Tay–Sachs and other genetic disorders can avoid marrying each other.
TREATMENT
Enzyme replacement therapy
Enzyme replacement therapy techniques have been investigated for lysosomal storage disorders, and could potentially
be used to treat Tay–Sachs as well. The goal would be to replace the nonfunctional enzyme, a process similar to insulin injections for diabetes.
However, in previous studies, the HEXA enzyme itself has been thought to be too large to pass through the specialized cell layer in the blood vessels
that forms the blood–brain barrier in humans. Researchers have also tried directly instilling the deficient enzyme hexosaminidase A into the cerebrospinal
fluid (CSF) which bathes the brain. However, intracerebral neurons seem unable to take up this physically large molecule efficiently even when it is
directly by them. Therefore, this approach to treatment of Tay–Sachs disease has also been ineffective so far.
Jacob Sheep Model
Tay–Sachs disease exists in Jacob sheep. The biochemical mechanism for this disease in the Jacob sheep is virtually identical to that in humans,
wherein diminished activity of hexosaminidase A results in increased concentrations of GM2 ganglioside in the affected animal. Sequencing of the
HEXA gene cDNA of affected Jacobs sheep reveal an identical number of nucleotides and exons as in the human HEXA gene, and 86% nucleotide sequence
identity. A missense mutation (G444R) was found in the HEXA cDNA of the affected sheep. This mutation is a single nucleotide change at the end of exon 11,
resulting in that exon's deletion (before translation) via splicing. The Tay– Sachs model provided by the Jacob sheep is the first to offer promise as
a means for gene therapy clinical trials, which may prove useful for disease treatment in humans.
Substrate Reduction Therapy
Other experimental methods being researched involve substrate reduction therapy, which attempts to use alternative enzymes to increase the brain's catabolism of GM2 gangliosides to a point where residual degradative activity is sufficient to prevent substrate accumulation. One experiment has demonstrated that using the enzyme sialidase allows the genetic defect to be effectively bypassed, and as a consequence, GM2 gangliosides are metabolized so that their levels become almost inconsequential. If a safe pharmacological treatment can be developed – one that increases expression of lysosomal sialidase in neurons without other toxicity – then this new form of therapy could essentially cure the disease Another metabolic therapy under investigation for Tay–Sachs disease uses miglustat. This drug is a reversible inhibitor of the enzyme glucosylceramide synthase, which catalyses the first step in synthesizing glucose-based glycosphingolipids like GM2 ganglioside.
Increasing β-hexosaminidase A activity
As Tay–Sachs disease is a deficiency of β-hexosaminidase A, by getting a substance that increases its activity, people affected will not be deteriorating as fast or not at all. While for infantile Tay–Sachs disease, there is no β-hexosaminidase A so then the treatment would be ineffective. However, for people affected by Late-Onset Tay–Sachs disease, they still have β-hexosaminidase A. The drug pyrimethamine has been shown to increase activity of β-hexosaminidase A.However, the increased levels of β-hexosaminidase A still fall far short of the desired "10% of normal HEXA", above which the phenotypic symptoms begin to disappear.
Cord Blood Transplant
This is a highly invasive procedure which involves destroying the patient's blood system with chemotherapy and administering cord blood. Of five people who had received the treatment as of 2008, two were still alive after five years and they still had a great deal of health problems. Critics point to its harsh nature, and that it is unapproved. It is also hard for the blood to cross the blood–brain barrier, as well as very expensive, as each unit of cord blood costs $25,000 and adults need many units of cord blood.